
Importing and merging files
Odds are that you will get raw data to work with that is in a CSV, sas7bdat, Excel’s xlsx file format, or something else. Stata can natively import many (but not all) file types. The simplest thing to do is use the –import– command then immediately save things as a temporary Stata .dta file and later merge them.
Importing commands differ by filetype. Type –help import– for details. But here are the 3 commands I use most frequently. These assume that the files are in the present working directory, which you can see by typing –pwd–. Remember to open Stata in Windows by double-clicking the .do file of interest in Windows explorer to set the folder that the .do file is sitting in as the working directory.
// CSV aka comma separated value, importing variable names
// from the first row
import delim using "NAME.csv", clear varnames(1)
save "name_csv.dta", replace // save as Stata dataset
// sas7bdat:
import sas using "NAME.sas7bdat", clear case(lower)
save "name_sas.dta", replace // save as Stata dataset
// Excel, MAKE SURE THAT YOU DON'T ALSO HAVE THE FILE
// OPEN IN EXCEL or it won't import it. This will import
// variable names from the first row.
import excel using "NAME.xlsx", clear firstrow.
save "name_xlsx.csv", replace // save as Stata datasetThere are lots of fancy settings within these commands.
To merge, simple merge all of your new .dta files using the –merge– command. This assumes that all files have a variable named “id” that uniquely identifies all rows and is preserved across files. eg:
use name_csv.dta
merge using name_sas.dta
drop _merge
merge using name_xlsx.dta
drop _mergeThe merge command generates a “_merge” variable that tells you where a specific variable came from. Review this variable and the output in Stata very closely. You need to drop the “_merge” variable before merging other datasets.
Getting the population, exposure, and outcome correct in your analytical dataset, and being able to come back and fix goofs later
Defining a study population, exposure variable, and outcome variable is a critical early step after determining your analysis plan. Most epidemiology projects come as a huge set of datasets, and you’ll probably need to join multiple files into one when defining your analytical population. Defining your analytical population is an easy place to make errors so you’ll want to have a specific script that you can come back and edit again if and when you find goofs.
For the love of Pete — Please generate your population, exposure, and outcome variables using a script so you can go back and reproduce these variables and fix any bugs you might find!
When you make these variables, you’ll likely need to combine several datasets. This will require mastery of importing datasets (if not in the native format for your statistical program) and combining datasets. For Stata, this means using –import– and –save– commands to bring everything over into Stata format, and then using –merge– commands to combine multiple datasets.
Make a variable for your population that is 0 (not included) or 1 (included)
One option in generating your dataset is to drop everyone who isn’t in your dataset. I recommend against dropping individuals who aren’t in your dataset. Instead, create a variable to define your population. Name it something simple like “included”, “primary population”, “group_a” or whatnot. If you will have multiple populations (say, one defined by prevalent hypertension using JNC7 vs. ACC/AHA 2017 hypertension thresholds), then you should have a variable for each addended with a simple way to tell them apart. Like “group_jnc7” and “group_aha2017”.
Useful code in R and Stata to do this:
- Count
- Generate and replace (Stata), mutate (R)
- Combine these with assigning single equals sign “=” (Stata & R, I say out loud “assign” when using this) and “<-" (R)
- use –if–, –and–, & –or– statements
- Tests of equality: >, =, <=, != (not), == ("equals exactly"), not single equal sign
Example Stata code to count # of people with diabetes, generate a variable for group_a and assign someone to group_a if they have diabetes.
count if diabetes==1
gen group_a=0
replace group_a=1 if diabetes==1
count if group_a==1 When creating a new variable from another variable, sometimes it’s helpful to start to generate a missing variable first (a dot) then replace a certain group as 0 and another as 1. This is especially helpful for strings. Strings must be in quotations, btw. Here’s an example of how to make a numeric variable from “Y” and “N” strings
gen diabetes = . // start with variable that's missing for all
replace diabetes = 0 if diab_string == "N"
replace diabetes = 1 if diab_string == "Y"Missing as positive infinity in Stata
Read up about missing values by typing –help missing–. In Stata, missing values are either a dot or a dot with a letter (e.g., “.” or “.a”). Mathematically, Stata considers a dot missing value to be positive infinity, and each dot and letter missing value to be even larger positive infinity, so: one billion trillion < . < .a < .b < .c and so on. Understanding that positive infinity is considered missing in Stata is critically important when using greater than statements, since anything greater than another value will include all missing values. So, imagine you had a population of 1 million people and only 100 of them were asked how many popsicles they have in their freezer and half had 0 to 10 and the other half had 11 to 20. If you try to make a variable called “popsicle_count” that is 1 for the lower half (0-10) and 2 for the higher half (11-20), you’d do something like this:
gen popsicle_count = . // everyone has a missing variable
replace popsicle_count=1 if popsicle <=10replace popsicle count = 2 if popsicle >10…you would the popsicle_count variable would have 25 people with a value of 1 and 999,975 with a value of 2. this is because the last line didn’t specify what to do with missing values. The easy workaround here is to use “& popsicle <." to specify that you wanted to include anyone with a value less than positive infinity, aka missing values, in the last line. The correct way of writing this would be:
gen popsicle_count = . // everyone has a missing variable
replace popsicle_count=1 if popsicle <=10
replace popsicle_count=2 if popsicle >10 & popsicle <. // NOTE!!This last line correctly ignores all variables when assigning a value of 2 since it applies the number of 2 to anything greater than 10 and less than positive infinity, aka anything less than a missing value. .
Here’s example R code to do the same (df=data frame).
nrow( df %>% filter(diabetes == 1) )
df = df %>% mutate(group_a = ifelse(diabetes == 1, 1, 0) )
Make an inclusion flowchart
These are essential charts in observational epidemiology. As you define your population, generate this sort of figure. Offshoots of the nodes define why folks are dropped from the subsequent node. Here’s how I approach this, folks might have different approaches:
- Node 1 is the overall population.
- Node 2 is individuals who you would not drop for baseline eligibility reasons (had prior event that discounts them or missing data to prevent assessment of their eligibility)
- Node 3 is individuals who you would not drop because you can assess them for necessary follow-up (incomplete follow-up, died before required follow-up time, missing data)
- Node 4 is individuals who you would not drop because they had all required exposure covariates (if looking at stroke by cholesterol level, people who all have cholesterol). This is your analytical population.

If you have two separate populations (eg, different hypertension populations by JNC7 or ACC/AHA 2017), you might opt to make two entirely separate figures. If you have slightly different populations because of multiple exposures (e.g., 3 different inflammatory biomarkers, but you have different missingness between the 3), you might have the last node fork off into different nodes, like this:
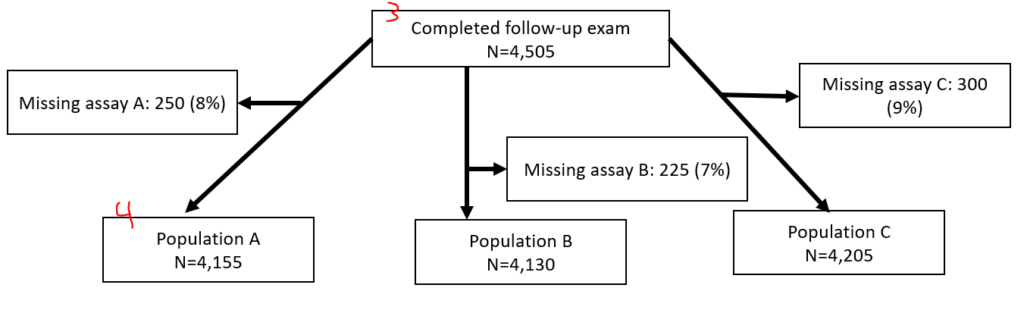
I generate these via text output in Stata then manually generate them in PowerPoint. To make these, I use a series of “sum” and “count” commands following along with “noisily display” commands all in a quietly loop. (Noisily overrides quietly for a single line. When debugging, you might want to hide the quietly loop.)
I also make an “include” variable that defines the analytical population(s) of interest.
You can display specific bits of data after a “sum” command, including r(N), which is the N of a sample. If you wonder what bits of data are available after a command like “sum if include==1”, type “return list”.
Example:
quietly {
gen include=1 // make a variable called "include" that is 1 for everyone
count if include ==1 // count the # of rows with an include variable equal to 1, this is everyone. It will save that value as r(N).
noisily display "Original study population, n= " r(N) // you can print this
//
// now lets print out the people we exclude
count if prevalent_htn_jnc7==1 // this will count the # with prevalent htn to be excluded
noisily display " --> Hypertension at baseline, n= " r(N)
count if prevalent_htn_missing==1
noisily display " --> Missing bp at baseline, n= " r(N)
//
// now we are going to replace the include variable as 0 for people missing the two things above
replace include = 0 if prevalent_htn_jnc7==1
replace include = 0 if prevalent_htn_missing==1
count if include==1
noisily display "normotensive at baseline, n= " r(N)
// and so on
}If you are using weighted data, this approach will differ slightly. First, you will have to svyset your data. Next, you will use “svy, subpop(IF COMMAND HERE): total [thing]”. Instead of using “return list”, you use “ereturn list” to see the bits that are saved. The weighted N is e(N_pop), for example.
svyset [pweight=sampleweight]
gen include=1
svy, subpop(if include==1): total include
ereturn list // notice e(b) is there, it's the beta from the prior estimation
noisily di "original study population, n= " e(b)[1,1]
// above prints out the first cell of the matrix e(b), hence the [1,1]
// type "matrix list e(b)" to see what's in that matrix.
// now figure out how many are excluded for missing a biomarker
svy, subpop(if include==1 & biomarker==.): total include
// now print it out, but since this uses sampling, it will not be a whole number. Print out a whole number with the %4.0f formatting code.
noisily di " --> missing biomarker, n= " %4.0f e(b)[1,1]
// now update the include to be 0 for missing biomarkers
// and display the count of the node print the node
replace include=0 if biomarker==.
svy, subpop(if include==1): total include
no di "not missing biomarker, n= " %4.0f e(b)[1,1]
// and so on
When you get to the end, you’ll have a variable called “include” that you will use in all of your later analyses to define your analytical population. Depending on your analysis, you might need to make a few different include variables. For example, we commonly run hypertension analyses using both jnc7 and acc/aha 2017 hypertension definitions, so I usually have an “include_jnc7” and also a separate “include_aha2017” variable.
Defining exposure and outcome
This seems simple, but define clearly what your exposure is and your outcome is. Each should have a simple 0 or 1 variable (if dichotomous) with an intuitive name. You might need 2 separate outcomes if you are using different definitions, like “incident_htn_jnc7” and “incident_htn_aha2017”.
Table 1
“Table 1” shows core features of the population by the exposure. Don’t include the outcome as a row, but include demographics and key risk factors/covariates for outcome (eg if CVD, then diabetes, blood pressure, cholesterol, etc.). Some folks include a 2nd column that presents the N for that row. Some folks also include a P-value comparison as a final row. I tend to generate the P value every time but only present it if the reviewers ask for it.
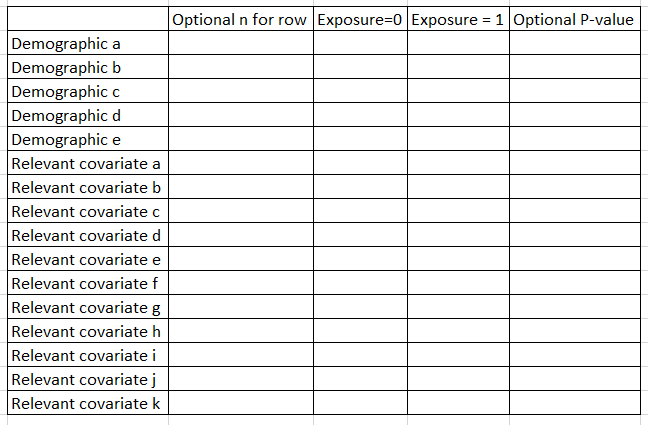
In Stata, I use the excellent table1_mc program to generate these, which you can read about here. If you are using p-weighted data, you can use this script that I wrote, here.
For R, I am told that gtsummary works well.
For lots more details on Table 1s, please continue to the next post, here.